ことの始まり
ホモ・プロテインcDNAバンクの中から、タンパク質間相互作用モチーフとして知られているWWドメイン(表1)を有する新規タンパク質cDNAクローンHP10345を選んで解析し、論文化しました(K99-3)。このタンパク質は核に存在し、SDS-PAGEで38kDaのバンドを生成したのでNpw38(Nuclear protein containing a WW domain with a molecular mass of 38 kDa)と命名しました。Npw38はほぼ同時に東京大の岡澤均博士のグループが報告したポリグルタミン結合タンパク質1(PQBP1)と同一でした[1]。その後、名称としてはPQBP1が採用されています。

ヒト遺伝子コレクションに含まれるPQBP1cDNA
論文に記載したcDNAクローンはヒト胃癌細胞由来ですが、ヒト遺伝子コレクションには網膜色素上皮細胞株ARPE-19由来のクローンARh10H01が一個含まれています(HP10345)。ベクターキャッピング法で得られたこのクローンの5’端は、ゲノムの配列にないGから始まっていませんが、転写開始点は胃癌細胞由来のものと同じなので完全長cDNAクローンであると思われます。
PQBP1の構造
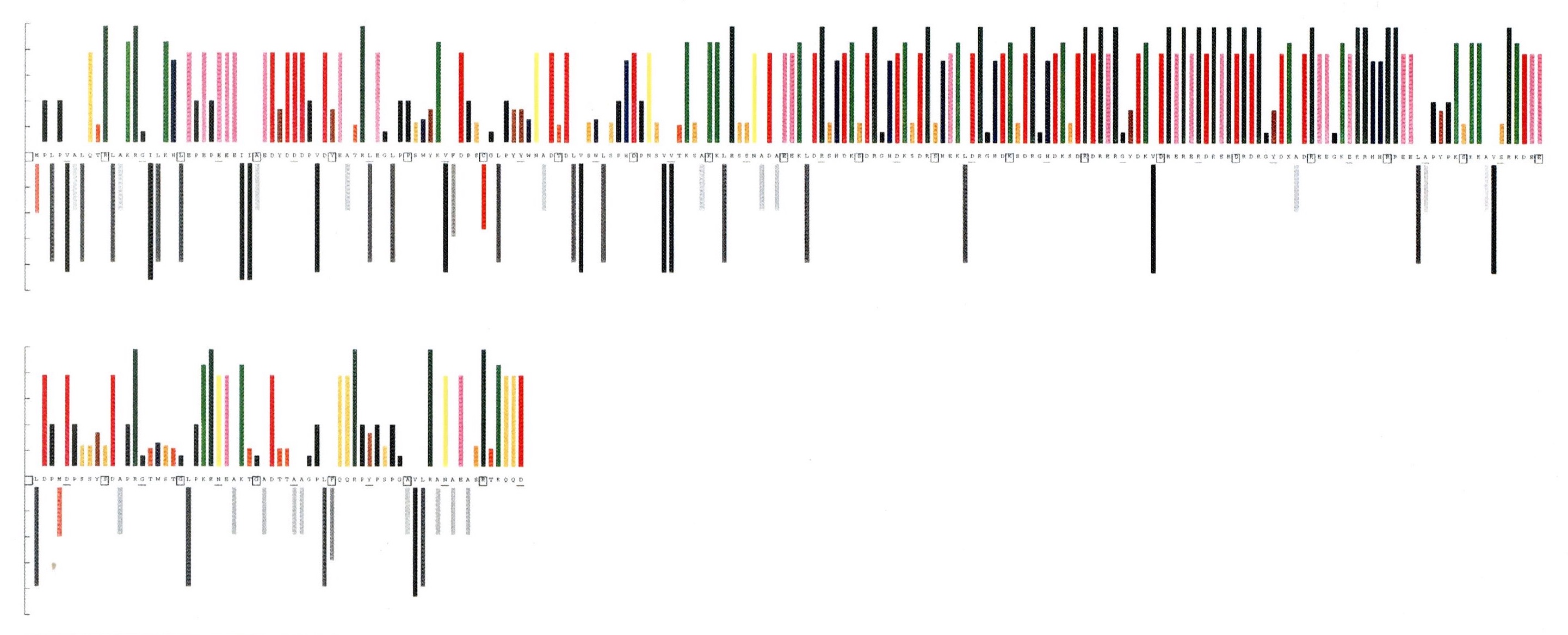
図1にPQBP1のドメイン構造を、図2にPQBP1のプロテオグラムを示します。PQBP1は21番目から41番目までのAspとGluを含む酸性領域、48番目から78番目までの2個のTrp、Gly、Tyr、Proが保存されたWWドメイン、101番目から170番目までの(Asp/Glu)(Lys/Arg)の繰り返しからなる極性アミノ酸リッチドメイン(PRD)、176番目から187番目までの核局在化シグナル(NLS)、それ以降のC末端ドメイン(CTD)という5つのドメインから構成されています。プロテオグラムを見ると、それぞれの特徴が一目でわかります。CTDでは8個のPro、3個のTyr、5個のGlyの存在が特徴的です。
図1 PQBP1のドメイン構造
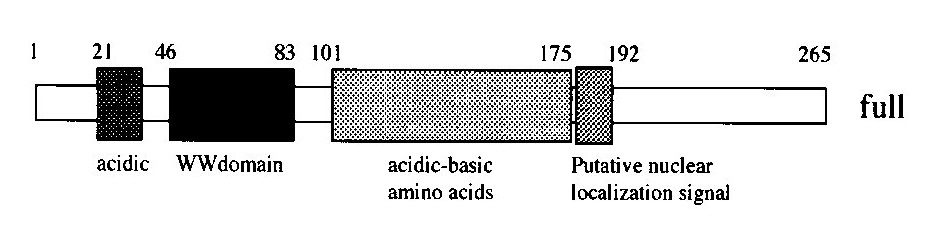
図2 PQBP1のプロテオグラム
The Human Protein AtlasでAlphafoldによるPQBP1の構造予測を見てみると、WWドメインは3本のアンチパラレルβシート構造を有しており、Val5-Lys13、Ala89-Asp104、Ser138-Glu183、Pro248-Lys262の各領域はαーヘリックス構造をとると予測しています。しかし、TakahashiらはNMR法を用いてPQBP1の構造解析を行い、WWドメイン以外の領域は特定の構造を取らず、フレキシブルな構造である天然変性タンパク質であることを明らかにしました[2]。
PQBP1の機能
PQBP1の機能に関しては、Tanakaらの総説[3]とWienchらの総説[4]によくまとめられています。PQBP1の機能を知るための方法の一つは、PQBP1と結合するタンパク質を明らかにすることです。PQBP1の各ドメインと結合するタンパク質がいくつか報告されているので、以下にまとめました。
- N端の酸性アミノ酸領域に、細胞内に入り込んだHIV-1のカプシドが結合する[5]。
- WWドメインは、スプライセオソーム複合体の一つであるWBP11[6]、RNAポリメラーゼIIのリン酸化されたCTD[7]、真核生物伸長因子2(eEF2)[8]、DNAセンサーである環状GMP-AMP合成酵素 (cGAS)[9]、毛様体形態形成に関与するダイナミン2[10]、外来性タウタンパク質[11]、鳥レオウイルスのp17[12]と結合する。
- PRDには転写因子Brn-2のポリグルタミン鎖が結合する[1]。
- NLSにはカリオフェリンβ2が結合し核内輸送を仲介する[13]。
- CTDにはスプライセオソームタンパク質U5-15KD[14]やHIV-1由来cDNA[9]が結合する。
なお、WWドメインに結合するタンパク質として最初に同定されたのは、我々が報告したNpwBP(後に名称はWBP11に統一)です(K99-7)[6]。また、我々はPRDにポリ(rG)RNAが結合することを示しました。
PQBP1は転写機構に関係するRNAポリメラーゼII、伸長因子であるeEF2、スプライセオソームの構成成分であるWBP11やU5-15KD、ポリ(rG)などに結合することから、転写・スプライシングにおいて重要な役割を果たしていることが示唆されます。また、HIV-1のカプシド、HIV-1由来cDNA、鳥レオウイルスのp17に結合することから、ウイルス感染に対する自然免疫においてウイルスセンサーとしての役割を有していることも示唆されます。
PQBP1が関与する疾患
Waragaiらは神経細胞転写因子Brn-2のポリグルタミン鎖に結合するタンパク質としてPQBP1を見つけました[1]。この研究はポリグルタミン病の原因を探る研究の一環として行われました。実際、PQBP1はポリグルタミン鎖が異常伸長した変異ataxin-1と結合し、その結果PQBP1に対するRNAポリメラーゼIIのリン酸化されたCTDの結合が増大し、RNAポリメラーゼIIの減少とそれに伴う転写の減少が、脊髄小脳変性症1型(Spinocerebellar ataxia type 1、SCA1) を引き起こすことが示唆されました[7]。
X連鎖性精神遅滞症候群の患者で、PQBP1のPRD内のアミノ酸配列DR/ERリピートの位置で、塩基配列AGリピートの伸長や短縮によるフレームシフトが起こり、PQBP1のNLSやCTDが欠失していることが見出されました[15]。ついで、X連鎖性精神遅滞症候群の一つであるGolabi-Ito-Hall症候群の患者のPQBP1のWWドメインの中にミスセンス変異p.(Tyr65Cys)が見つかりました[16]。この変異PQBP1はそのパートナーであるWBP11と結合できなくなり、患者由来のリンパ球芽細胞内でのスプライシング効率が減少することが見出されました[17]。さらに、Renpenning症候群の患者で、PQBP1のCTDに見つかったミスセンス変異p.(Pro244Leu)によって、PQBP1とスプライセオソームタンパク質U5-15KDとの結合が阻害されれることが示されました[13]。Renpenning症候群に似た症状を示す患者では、PQBP1遺伝子を含む領域の重複が認められたことから、PQBP1の過剰発現が病気の原因である可能性が示唆されました[18]。以上のように、PQBP1のミスセンス変異、塩基の挿入や欠失によるフレームシフト、過剰発現などがX連鎖性精神遅滞症候群の原因であることが示唆されています。
ミクログリア内でPQBP1が外因性のタウタンパク質と直接相互作用し、環状GMP-AMP合成酵素(cGAS)-インターフェロン遺伝子刺激因子(STING)経路を活性化することで自然免疫応答を引き起こすことが報告されました[11]。タウタンパク質による脳内炎症がアルツハイマー病と関連していると考えられていることから、これは興味深い結果です。